Нужно почти $12 миллионов. Четыре мальчика из Беларуси ждут один из самых дорогих уколов в мире
В прошлом году многие белорусы пристально следили за самым крупным благотворительным сбором в истории страны. Тогда собирали $2,9 миллиона на спасительный укол для Артема Подъяловского — мальчику поставили страшный диагноз «миопатия Дюшенна». Первый препарат от заболевания — тот самый чрезвычайно дорогой Elevidys — появился только в 2023 году. К счастью, огромную сумму собрали, и Артему уже сделали инъекцию. Но болеет, увы, не только он. Прямо сейчас в Беларуси открыты четыре аналогичных сбора.
Что это за болезнь?
Миопатию Дюшенна называют генетическим убийцей мальчиков номер один в мире (среди девочек болезнь встречается один раз на 50 миллионов, а среди мальчиков — раз на 3,5—5 тысяч). Причина болезни — в неправильной работе гена дистрофина, который не производит достаточно белка, необходимого для работы и развития мышц.
Среди редких генетических заболеваний мышечная дистрофия Дюшенна является одним из наиболее распространенных. С прошлого года ООН объявила 7 сентября Всемирным днем распространения информации об этой болезни.
Обычно диагноз ставят ближе к четырем годам — родители замечают, что ребенок испытывает трудности с определенными движениями. Детям обычно сложно бегать, прыгать, подниматься по ступенькам, появляется характерная «утиная походка». Даже с учетом поддерживающей терапии ближе к подростковому возрасту большинству становится нужна инвалидная коляска. После 13 лет проявляются проблемы с сердцем и легкими. По статистике, к 20 годам большинство пациентов с этим диагнозом умирают.
Изначально для Elevidys действовали жесткие ограничения: его вводили пациентам только в возрасте 4—5 лет. Сейчас возрастные рамки сняли.
«Сын спросил: „Мама, а я умру?“»
Именно благодаря изменению правил совсем недавно начать сбор смогли близкие Артема Корбаля из Мозыря — мальчику уже исполнилось 10 лет. Мы поговорили с мамой Артема — Мариной.
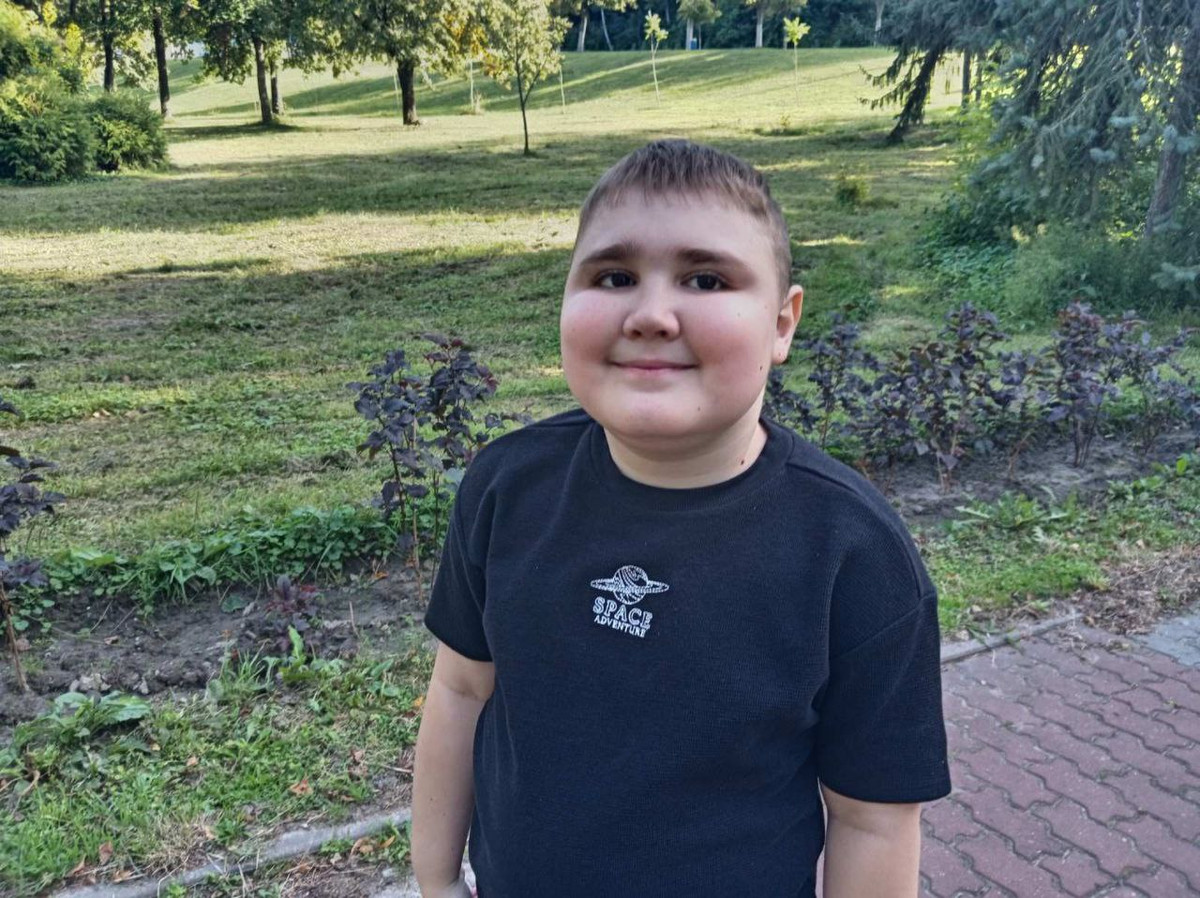
— У Артема была задержка речи, увеличены икроножные мышцы, он очень быстро уставал. И мы решили лечь на обследование в больницу. Тогда, в 4 года, и узнали его диагноз — миодистрофия Дюшенна, или МДД, — рассказала женщина.
До этого возраста болезнь не давала о себе знать — развивался малыш согласно нормам. Взрослые замечали легкие проблемы с координацией движений, но не подозревали в этом серьезное заболевание, списывали на индивидуальные особенности.
На тот момент даже речи не было о лечении. Все, что могли предложить врачи, это поддерживающая терапия. Но даже она подходила не для всех видов мутаций. Артему назначили гормоны — с тех пор он принимает их на постоянной основе: они замедляют прогрессирование болезни.
Заболевание генетическое. Мутация может передаться по материнской линии или случиться на этапе формирования организма самого ребенка. Марина выяснила, что она сама является носительницей, а вот ее мама — нет. То есть поломка произошла на ней, хоть на ее собственном здоровье никак не отразилась. Но из-за этого могут возникнуть трудности с рождением других детей, особенно мальчиков.
Сейчас Артем учится в 4-м классе обычной общеобразовательной школы. Родители всегда отвозят и забирают его, но по самому зданию он перемещается сам.

— Проблематично, конечно, и по лестнице подниматься, и долго ходить. Нужно преодолевать только один этаж — хоть и медленно, но он справляется. В прошлом году он еще мог подняться без опоры на перила или мою руку, а сейчас уже нет. Но все равно в первом и втором классе было куда сложнее. Маленькие дети и толкали, и обидеть могли. Но сейчас все повзрослели и только поддерживают его. Часто слышу: «Темка, давай рюкзак тебе поднесем». Очень хорошо к нему относятся, — говорит Марина.

Когда появилась информация о том, что Elevidys прошел сертификацию, для семьи это ничего не изменило — Артем уже не проходил по возрасту для лечения. Но как только стало понятно, что правила изменились и главное, чтобы ребенок не утратил способность ходить, семья обратилась в дубайскую клинику за консультацией. Там подтвердили: на этом этапе Артему еще можно помочь. В середине августа родители открыли сбор, пока удалось собрать меньше процента от нужной суммы.
— Конечно, очень тяжело. Четыре огромных сбора только с нашим диагнозом, а ведь есть еще СМА… Сумма неподъемная для обычных людей. Казалось бы, хотя бы миллион — это можно осознать. А тут $2 900 000. Но, если честно, внутренняя уверенность есть, что соберем. Не на 100%, но есть. Пока существует возможность спасти сына, мы будем делать все, — уверяет собеседница.
Марина добавляет, что о своей болезни Артем прекрасно знает, но эту тему затрагивает редко. Только несколько раз задавал вопрос: «Мама, а я умру?» В семье стараются жить так, чтобы не быть в плену диагноза, — по мере возможности ведут обычную жизнь.

Например, Артем просто обожает футбол. Его кумир — Криштиану Роналду. Кажется, даже посреди ночи он без труда назовет любой факт про любимого футболиста. Сам пытается учиться набивать мяч и регулярно выходит на местный стадион, чтобы поиграть с папой в футбол.
— Мой муж — не родной отец Артема. Но как только стало известно о диагнозе, он сделал мне предложение и пообещал поддерживать нашу семью до последнего, а если понадобится, даже жизнь отдать за нас, — отмечает Марина. — У них с Артемом особая связь. Они просто обожают друг друга, и Артем безоговорочно считает его своим отцом.

Сам Артем очень активный, смешной, радостный ребенок. Он у нас вообще не плачет. А еще нас самих заряжает.
- Информация о том, как можно помочь Артему, доступна здесь.
«Уже был момент, когда он не смог сам встать с кровати»
Как упоминала Марина, сейчас открыты еще три сбора на укол. О Ване Стеценко мы рассказывали в конце июня. За это время состояние 9-летнего мальчика резко ухудшилось. На то, чтобы успеть ввести укол, остается все меньше времени. На данный момент собрано около 60% от всей суммы, но до закрытия сбора нужно еще больше 1,17 миллиона.



В мае уже был момент, когда Ваня не смог сам встать с кровати. Тогда мальчику удалось помочь. Но без терапии рано или поздно наступит момент, когда он больше не сможет двигаться самостоятельно.
- Информация о том, как можно помочь Ване, доступна здесь.
На лечение собирают и для Егора Ярмоловича из Гродно. В конце сентября мальчику исполнится 9 лет, сейчас он учится во 2-м классе. На данный момент собрано 4,5%.
Посмотреть эту публикацию в Instagram
«Изначально у Егора был просто пониженный тонус мышц, он ходил на массажи и лечебную гимнастику. Позже других детей начал ползать, ходить, говорить стал после 3 лет. В остальном развитие было в норме. В 2 года он знал все цвета, фигуры и буквы алфавита. Когда начал ходить, часто спотыкался даже на ровном месте, по ступенькам поднимался с трудом. В 4 года врач начала подозревать миопатию. Но первый анализ диагноз не подтвердил, и мы успокоились — зря. Еще один анализ показал миопатию Дюшенна. Сейчас Егор ходит, старается делать все самостоятельно, учится в обычной школе во 2-м классе. Но в последнее время все чаще стал жаловаться на то, что у него болят ножки. А значит, болезнь прогрессирует», — рассказала мама.
- Информация о том, как можно помочь Егору, доступна здесь.
Даниилу Дуку из Новолукомля 6 лет. С каждым днем ему все сложнее выполнять обычные действия, поэтому родители решили пока не отдавать мальчика в школу. На 5-й этаж дома, где живет семья, самостоятельно подняться Даня уже не может. О диагнозе родители узнали только в начале этого лета, а сбор открыли в середине августа. К началу сентября собрано чуть больше 2% от всей суммы — $63 000.
Посмотреть эту публикацию в Instagram
- Информация о том, как можно помочь Дане, доступна здесь.
Есть о чем рассказать? Пишите в наш телеграм-бот. Это анонимно и быстро
Перепечатка текста и фотографий Onlíner без разрешения редакции запрещена. ga@onliner.by